Skrátená informácia o lieku
FEIBA 100 U/ml prášok a rozpúšťadlo na infúzny roztok.
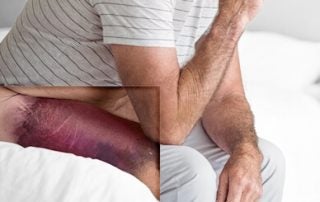
Zloženie lieku: Antiinhibičný komplex koagulačných faktorov (Factor Eight Inhibitor Bypassing Activity, FEIBA), 1 ml obsahuje 100 U (jednotiek) antiinhibičného komplexu koagulačných faktorov. Lieková forma: Prášok a rozpúšťadlo na infúzny roztok. Biely, takmer biely alebo slabozelený prášok. Pripravený roztok má hodnotu pH medzi 6,5 a 7,3. Indikácie: Liečba a profylaxia krvácania u pacientov s hemofíliou A s inhibítormi, liečba krvácania u pacientov s hemofíliou B s inhibítormi, liečba a profylaxia krvácania u nehemofilikov so získanými inhibítormi faktora VIII, profylaxia v prípade chirurgických zákrokov u pacientov s hemofíliou A s inhibítorm.FEIBA je vhodná pre všetky vekové kategórie.* Kontraindikácie: Ak sú k dispozícii terapeutické alternatívy, FEIBA sa nesmie použiť v nasledujúcich situáciách: Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok, diseminovaná intravaskulárna koagulácia (DIK), akútna trombóza alebo embólia (vrátane infarktu myokardu). Osobitné upozornenia: Reakcie z precitlivenosti: FEIBA môže vyvolať reakcie z precitlivenosti alergického typu, ako je urtikária, angioedém, gastrointestinálne prejavy, bronchospazmus a hypotenzia; tieto reakcie môžu byť závažné a môžu byť systémové. Pacienti majú byť informovaní o včasných prejavoch reakcií z precitlivenosti, napríklad o erytéme, kožnej vyrážke, generalizovanej urtikárii, prurite, dýchacích ťažkostiach/dýchavici, tlaku na hrudníku, celkovej indispozícii, závratoch a poklese krvného tlaku až po alergický šok. Pri prvých prejavoch a príznakoch hypersenzitívnej reakcie/reakcie na infúziu sa podávanie FEIBY musí zastaviť a začať náležitá liečba. Trombotické a trombembolické príhody: Počas liečby FEIBOU sa vyskytli trombotické a trombembolické príhody, ako je diseminovaná intravaskulárna koagulácia (DIK), venózna trombóza, pľúcna embólia, infarkt myokardu a cievna mozgová príhoda. FEIBA sa má používať obzvlášť opatrne a len vtedy, ak neexistujú žiadne terapeutické alternatívy u pacientov so zvýšeným rizikom trombembolických komplikácií. Trombotická mikroangiopatia (TMA) nebola hlásená v klinických štúdiách s FEIBOU. Prípady TMA boli hlásené v klinickom skúšaní s emicizumabom, keď pacienti dostávali FEIBU ako súčasť liečebného režimu pri epizódach akútneho krvácania. Pri prvých prejavoch alebo príznakoch trombotických alebo trombembolických príhod sa musí podávanie infúzie okamžite ukončiť a musia sa začať náležité diagnostické a liečebné opatrenia. Rôzna odpoveď na „bypassové“ lieky: Vzhľadom na faktory špecifické pre daného pacienta sa môže jeho odpoveď na „bypassové“ lieky líšiť a pri danom krvácavom stave môže pacient na jeden typ reagovať a na druhý nie. Ak nedostatočne odpovedá na jeden typ „bypassového“ lieku, má sa zvážiť podanie iného. Anamnestické odpovede: Podávanie FEIBY pacientom s inhibítormi môže vyvolať úvodný „anamnestický“ vzostup hladín inhibítora. S pokračujúcim podávaním FEIBY môže inhibítor po čase klesnúť. Klinické a publikované údaje naznačujú, že účinnosť FEIBY nie je znížená. Ovplyvnenie laboratórnych vyšetrení: Po podaní vysokých dávok FEIBY môže prechodný vzostup pasívne prenesených protilátok proti povrchovým antigénom vírusu hepatitídy B zapríčiniť chybnú pozitívnu interpretáciu výsledkov sérologického vyšetrenia. FEIBA obsahuje izohemaglutiníny krvných skupín (anti-A a anti-B). Pasívny prenos protilátok proti antigénom erytrocytov, napríklad A, B, D, môže interferovať s niektorými sérologickými testami na protilátky červených krviniek, ako je antiglobulínový test (Coombsov test). Sodík: 500 U
FEIBA obsahuje približne 40 mg sodíka na jednu injekčnú liekovku, čo zodpovedá 2% WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu. 1 000 U FEIBA obsahuje približne 80 mg sodíka na jednu injekčnú liekovku, čo zodpovedá 4 % WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu. 2 500 U FEIBA obsahuje približne 200 mg sodíka na jednu injekčnú liekovku, čo zodpovedá 10 % WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu. Interakcie: Pri súbežnom podávaní FEIBY spolu so systémovými antifibrinolytikami, ako je kyselina tranexámová a aminokaprónová, je potrebné zvážiť možnosť trombembolickej príhody. Preto sa antifibrinolytiká nemajú podávať približne 6 až 12 hodín po podaní FEIBY. Pri súbežnom podaní rFVIIa sa podľa dostupných údajov in vitro v a klinických pozorovaní nemôžu vylúčiť potenciálne liekové interakcie (čo môže viesť k nežiaducim účinkom, ako sú trombembolické príhody). Počas dvoch klinických skúšaní s emicizumabom dostalo 23 účastníkov, ktorí dostávali profylaxiu emicizumabom, aj FEIBU na liečbu 78 akútnych krvácaní. Zo 78 krvácaní bolo 59 zvládnutých priemernou dennou dávkou ≤ 100 U/kg/deň počas ≤ 2 dní bez komplikácií TMA. 19 zo 78 krvácaní bolo zvládnutých s dávkou > 100 U/kg/deň počas > 1 dňa s komplikáciou TMA, ktorá sa vyskytla u 3 pacientov (z toho 2 pacienti dostali aj rFVIIa pre tú istú príhodu krvácania).* Nežiaduce účinky: FEIBA môže vyvolať reakcie z precitlivenosti alergického typu, ktoré zahŕňajú žihľavku, angioedém, gastrointestinálne prejavy, bronchospazmus a pokles krvného tlaku; tieto reakcie môžu byť závažné a môžu byť systémové (napr. anafylaxia s urtikáriou a angioedémom, bronchospazmus a obehový šok). Dávkovanie a spôsob podávania: Dávkovanie a trvanie liečby závisí od závažnosti hemostatickej poruchy, lokalizácie a rozsahu krvácania a klinického stavu pacienta. Dávkovanie a frekvencia podávania sa vždy riadi podľa individuálneho klinického účinku u každého pacienta. Všeobecne sa odporúča podávať 50 – 100 U FEIBA/kg telesnej hmotnosti; jednorazová dávka nemá prekročiť 100 U/kg telesnej hmotnosti a maximálna denná dávka 200 U/kg telesnej hmotnosti, pokiaľ si závažnosť krvácania neoprávňuje a neodôvodňuje použitie vyšších dávok. Pediatrická populácia: Skúsenosti u detí mladších ako 6 rokov sú obmedzené; môže sa použiť rovnaký dávkovací režim ako u dospelých, upravený podľa klinického stavu dieťaťa. Spontánne krvácanie: Krvácanie do kĺbov, svalov a mäkkých tkanív - Pri miernom až stredne závažnom krvácaní sa odporúča dávka 50 až 75 U/kg telesnej hmotnosti v 12-hodinových intervaloch. V liečbe sa má pokračovať, pokým sa neobjavia jasné znaky klinického zlepšenia, ako napríklad zmiernenie bolesti, zmenšenie opuchu alebo zvýšenie pohyblivosti kĺbu. Pri závažnom krvácaní do svalov a mäkkých tkanív, akým je krvácanie do retroperitoneálneho priestoru, sa odporúča dávka 100 U/kg telesnej hmotnosti v 12-hodinových intervaloch. Krvácanie slizníc - Odporúča sa dávka 50 U/kg telesnej hmotnosti každých 6 hodín za starostlivého sledovania pacienta (vizuálna kontrola krvácania, opakované stanovenie hodnoty hematokritu). Ak nedôjde k zastaveniu krvácania, dávka sa môže zvýšiť na 100 U/kg telesnej hmotnosti, nesmie sa však prekročiť denná dávka 200 U/kg telesnej hmotnosti. Iné závažné krvácania - Pri závažnom krvácaní, akým je krvácanie do CNS, sa odporúča dávka 100 U/kg telesnej hmotnosti v 12-hodinových intervaloch. V jednotlivých prípadoch sa FEIBA môže podávať v 6-hodinových intervaloch, pokým sa nedosiahne jasné zlepšenie klinického stavu. (nesmie sa prekročiť maximálna denná dávka 200 U/kg telesnej hmotnosti). Chirurgické zákroky: Pri chirurgických zákrokoch možno predoperačne podať úvodnú dávku 100 U/kg telesnej hmotnosti a ďalšiu dávku 50 – 100 U/kg telesnej hmotnosti možno podať po 6 – 12 hodinách. Ako pooperačnú udržiavaciu dávku možno podávať 50 – 100 U/kg telesnej hmotnosti v 6 – 12-hodinových intervaloch; dávkovanie, dávkovacie intervaly a trvanie peri- a pooperačnej liečby sa riadia chirurgickým zákrokom, celkovým stavom pacienta a klinickou účinnosťou v každom jednotlivom prípade. (Nesmie sa prekročiť maximálna denná dávka 200 U/kg telesnej hmotnosti!) Profylaxia u pacientov s hemofíliou A s inhibítormi: Profylaxia krvácania u pacientov s vysokým titrom inhibítora a s častým krvácaním, u ktorých nebola úspešná imunotolerančná liečba (Immune Tolerance Induction, ITI) alebo sa o nej neuvažuje: Odporúča sa dávka 70 – 100 U/ kg telesnej hmotnosti každý druhý deň. Ak je to nevyhnutné, dávka sa môže zvýšiť až na 100 U/ kg telesnej hmotnosti denne alebo sa môže postupne znížiť. Profylaxia krvácania u pacientov s vysokým titrom inhibítora, ktorí podstupujú imunotolerančnú liečbu (ITI): FEIBA sa môže podávať súbežne s podávaním faktora VIII, v rozmedzí dávky od 50 do 100 U/kg telesnej hmotnosti dvakrát denne, až kým nedôjde k poklesu titra inhibítora faktora VIII na < 2 BU (Bethesda jednotka). Spôsob podávania: Podávajte pomalou intravenóznou infúziou. Rýchlosť podávania FEIBY je 2 U/kg telesnej hmotnosti za minútu. U pacientov, ktorí dobre tolerujú rýchlosť podávania infúzie 2 U/kg telesnej hmotnosti za minútu, sa môže v nasledujúcich infúziách zvýšiť rýchlosť podávania až na maximum 10 U/kg telesnej hmotnosti za minútu. Fertilita, gravidita, laktácia: Gravidita: Nie sú žiadne dostupné údaje o použití FEIBY u gravidných žien. Lekári musia zvážiť potenciálne riziká a predpísať FEIBU iba v nevyhnutných prípadoch, pričom treba vziať do úvahy, že gravidita je spojená so zvýšeným rizikom trombembolických príhod a že niektoré komplikácie gravidity sú spojené so zvýšeným rizikom DIK.* Dojčenie: Nie sú k dispozícii žiadne dostatočné údaje o použití FEIBY počas dojčenia. Koagulačné faktory sú veľké molekuly bielkovín, preto je ich množstvo v materskom mlieku pravdepodobne veľmi nízke. Keďže však nie sú k dispozícii žiadne údaje, lekári musia zvážiť potenciálne riziká a predpísať FEIBU iba v nevyhnutných prípadoch, pričom treba vziať do úvahy, že popôrodné obdobie je spojené so zvýšeným rizikom trombembolických príhod.* Fertilita: Reprodukčné štúdie na zvieratách s FEIBOU neboli zrealizované a ani účinky FEIBY na fertilitu neboli v kontrolovaných klinických štúdiách stanovené. Držiteľ rozhodnutia o registrácii: Baxalta Innovations GmbH Industriestrasse 67, 1221 Viedeň, Rakúsko. Registračné číslo: 16/0109/24-S. Výdaj lieku je viazaný na lekársky predpis. Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na Štátny ústav pre kontrolu liečiv, Sekcia klinického skúšania liekov a farmakovigilancie, Kvetná ul. 11, SK-825 08 Bratislava, Tel: + 421 2 507 01 206, e-mail: neziaduce.ucinky@sukl.sk. Tlačivo na hlásenie nežiaduceho účinku je na webovej stránke www.sukl.sk v časti Bezpečnosť liekov/Hlásenie o nežiaducich účinkoch. Formulár na elektronické podávanie hlásení: https://portal.sukl.sk/eskadra/. Hlásením vedľajších účinkov môžete prispieť k získaniu ďalších informácií o bezpečnosti tohto lieku. Hlásenie akékoľvek podozrenia na nežiaduce reakcie sa vyžaduje aj spoločnosti Takeda emailom na AE.SVK @takeda.com.
Dátum poslednej revízie textu: 22. 11. 2024.* Dátum vypracovania aktualizácie reklamy: 03/2025. Pred predpísaním lieku sa oboznámte s úplným znením Súhrnu charakteristických vlastností lieku uvedenom na stránke ŠÚKLu, resp. EMA, alebo dostupnom u lokálneho zástupcu: Takeda Pharmaceuticals Slovakia s.r.o., Svätoplukova II., 18892/2 A, 821 08 Bratislava, Slovenská republika, tel.: +421 2 206 026 00.
*Všimnite si prosím zmenu(y) v súhrne charakteristických vlastností lieku.